
魏先生60岁了,据他自己回忆,20多岁的时候就被诊断为“多囊肝多囊肾”,40多岁的时候因肾功能衰竭先后行透析治疗和肾移植。现在反复出现腹水来住院。
此外,有一个重要的家族史:患者的弟弟也患有多囊肾。
根据临床判断,魏先生很有可能患的是一种叫“常染色体显性遗传多囊肾病(ADPKD)”,是一种常见的单基因常染色体显性遗传病,遗憾的是魏先生因个人原因拒绝完善基因检测。
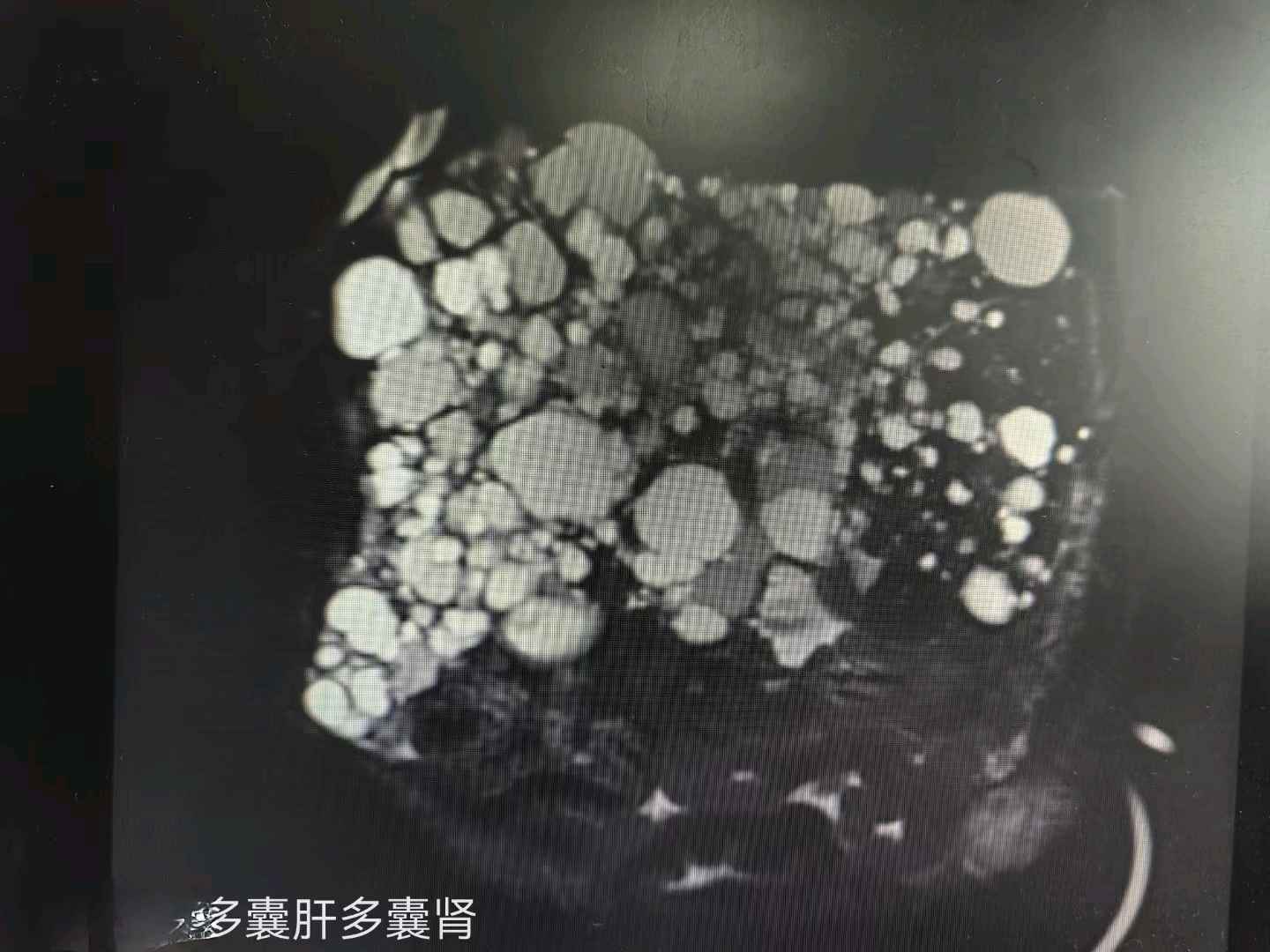 上图:魏先生的肝脏磁共振
上图:魏先生的肝脏磁共振常染色体显性遗传多囊肾病(ADPKD),以双侧肾囊肿、肝囊肿、颅内动脉瘤和二尖瓣脱垂风险增加为特征,外显率近100%,发病率为1/4000-1/1000,这个发病率不低了,目前我国约有150万患者。
发病年龄多在40~60岁,故又称为“成人型多囊肾病”。其中,85%的ADPKD患者是由PKD1基因突变导致,15%的ADPKD患者由PKD2基因突变导致,此外,近期还发现了一些少见的微小基因如GANAB基因。
PKD1基因位于染色体16P13,是比较常见的致病基因,而且导致更加严重的表型,这一基因编码膜内在蛋白,如pplucystin1,它涉及细胞与细胞之间以及细胞与基质之间的相互作用。PKD2基因位于染色体4q21-q23,比较少见,它编码蛋白产物polycystin2,用于调控管道形态和功能。
常染色体隐性遗传多囊肾(ARPKD)与之相反,是青少年多见,被称为“婴儿型多囊肾”,致病基因也不同。发病率较ADPKD更低。
ADPKD导致肝内出现大量的胆管错构瘤(VMC),并且呈不同程度的扩张和囊性改变。这些囊性改变一般只是在形态上增大,而且铆钉在肝脏实质内。相反,ARPKD导致的肝脏囊性改变保持与胆道系统的联系。尽管Caroli病与ARPKD相关,但一些病理报告显示其与ADPKD也有关。
而多囊肝(PLD)以前也被认为是ADPKD的一种,但现在发现PLD与ADPKD是不同的疾病:PLD可以不合并多囊肾,而且致病基因不同(虽然二者都是常染色体显性遗传),目前已有多个基因位点被证实:PRKCSH(约占ADPLD病例的20%)和SEC63(约占ADPLD病例的15%)等,它们编码的是hepatocystin蛋白。
ADPKD对肾脏的影响更大,大约一半的患者在 40~70 岁期间进展为终末期肾病(ESRD)。因此这类病人在肾内科更常见。大多数ADPKD儿童患者无症状,部分表现为高血压或肉眼血尿。有些患者出生即有囊肿,只是较小,不易查出,20岁以前一般不易发现。患者在30~40岁,囊肿将有一较快的生长期。患者进入40岁以后,囊肿会有进一步长大,患者出现较多的临床症状,如腰痛、蛋白尿、血尿、高血压和肾功能进行性恶化等。有些患者出现囊肿破裂出血和感染。最后患者会逐渐进展至尿毒症。
多囊肝是ADPKD的常见肾外表现,一般无明显症状,且发生比肾囊肿更晚,据统计60岁以后有70%的患者可发现多囊肝,且发病率并不与肾囊肿的严重程度相平行。肝大是患者早期常见症状,囊肿累及肝脏但并不改变其功能,因此本病例的魏先生也未见明显肝功能异常。但是肝脏囊肿过大引发的压迫症状明显时,需要外科处理。减少囊肿和肝体积的治疗方法,可行肝囊肿穿刺硬化、腹腔镜下或开放手术囊肿去顶减压术,甚至行肝部分切除术或肝移植术。
ADPKD中动脉瘤破裂的发生率是一般人群的5倍,蛛网膜下腔出血风险增加,这是ADPKD最可怕的并发症,应提高警惕。
ADPKD呈常染色体显性遗传,约95%的患者有家族史,约5%的患者是由新发突变引起,如果父母一方患病且携带致病/疑似致病变异,其每个子女的理论患病风险为50%,建议在生育前进行产前检测及遗传咨询。
