深圳的石先生,今年30多岁了。
1个月前来到我的门诊:付医生,我从生下来就身目黄染,一直没重视,10多年前开始体检,每年体检胆红素都高,看了好多家医院,一直没搞清楚,今天让您帮我看一下。
我看了看石先生最近的体检单,总胆红素 153umol/L(正常值5.1-20.5),间接胆红素146umol/L(正常值0-14.2)。其他检查比如转氨酶、血常规、肝脏彩超等都是正常的。
我问石先生:您平时有什么不舒服么?胆红素水平会有波动吗?
石先生说:都挺好的,没什么不舒服,除了尿黄,其他方面比如胃口、精神都跟平常人一样。黄疸基本没有消失过,总胆红素一直处于150左右。
我又问:家人有类似病史么
石先生告诉我:父亲也有黄疸病史,跟我的情况差不多。
我告诉他:如果排除了溶血性黄疸,您这种情况高度怀疑一类叫“先天性非溶血黄疸”的疾病,是由于基因突变导致胆红素代谢异常。
石先生说:我也在网上查过很多资料,听说过这种病。要怎样才能确诊呢?
我告诉他:可以通过检测基因来确诊,就是费用偏高,需要自费。
石先生说:我已经糊里糊涂过了30多年了,这次一定要搞清楚才放心,那就做吧。
通过排查了常见的溶血性疾病和引起黄疸的肝病,同时也抽血送了先天性非溶血性黄疸的基因检测。
过完国庆节假期,石先生的结果出来了。

结果提示UGT1A1基因的复合杂合突变,结合石先生的黄疸一直比较高,高度怀疑患者是先天性非溶血性黄疸中的Crigler-Najjar综合征Ⅱ型。经请教中山三院的肝脏罕见病专家李新华教授,也同意目前的诊断。我给石先生打电话汇报了这一结果,石先生也终于放下了心里这块装了30多年的石头。
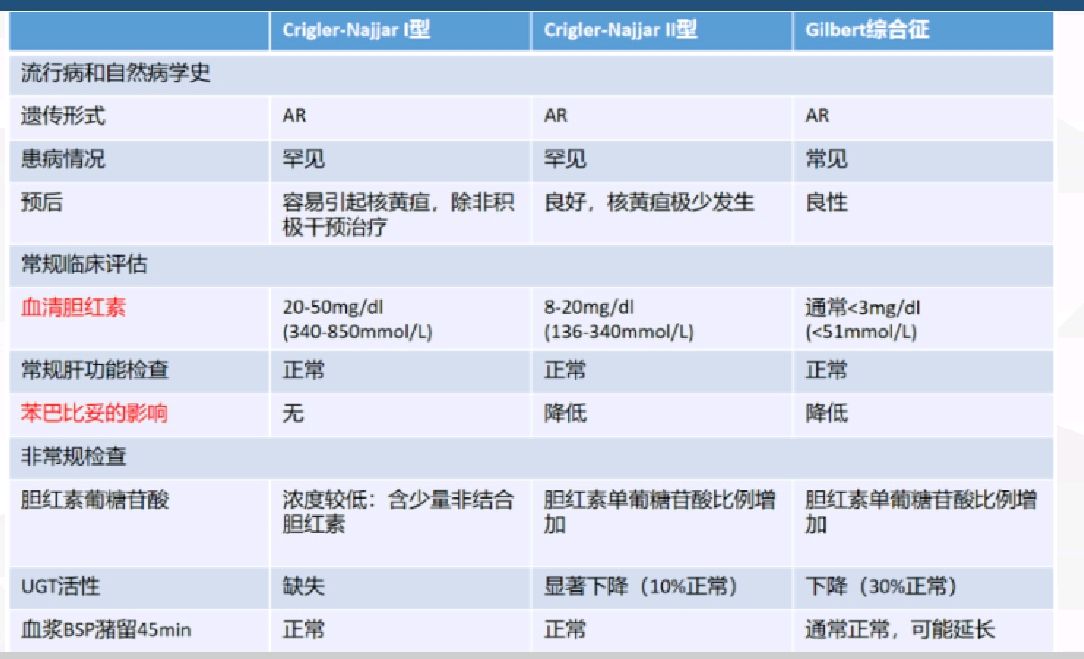
黄疸是指血浆胆红素中浓度升高(>34.2mmol/L或2mg/dL)沉积于组织中,引起巩膜、皮肤、黏膜及其他组织和体液发生黄染的现象。在临床,黄疸的病因学分类可分为以下四种:溶血性黄疸、肝细胞性黄疸、胆汁淤积性黄疸(梗阻性黄疸)、先天性非溶血性黄疸。
先天性非溶血性黄疸是黄疸性疾病中的一大类,是由于先天酶缺陷所致肝细胞对胆红素的摄取、结合及排泄障碍引起的黄疸。临床上相对罕见,多属遗传代谢类肝病。
主要包括两大类:(1)以非结合胆红素升高为主的疾病,包括Gilbert综合征和Crigler-Najjar综合征(Ⅰ型和Ⅱ型);(2)以结合胆红素升高为主的疾病,包括Dubin-Johnson综合征、Rotor综合征、良性复发性肝内胆汁淤积症和家族性进展性肝内胆汁淤积症。
本文主要介绍以间接胆红素升高为主的三种疾病,Gilbert综合征、Crigler-Najjar综合征Ⅰ型和Crigler-Najjar综合征Ⅱ型。
这三种疾病的突变基因都是在尿苷二磷酸葡萄糖醛酸转移酶(uridine glucuronosyl transferase, UGT)1A1(UGT1A1)基因的突变,UGT1A1酶是分布于肝脏内质网,参与胆红素代谢,负责促进游离胆红素和葡萄糖醛酸结合,形成葡萄糖醛酸胆红素,即促进游离胆红素转化为结合胆红素。当UGT1A1基因出现突变,导致UGT1A1酶活性受损,游离胆红素不能正常转化为结合胆红素,使游离胆红素蓄积,形成高非结合胆红素血症。不同的UGT1A1基因突变,形成不同程度的UGT1A1酶活性缺失,导致不同程度的高胆红素血症。
简而言之,三者是相同发病机制、相似病理过程,不同严重程度的UGT1A1异常谱系疾病。
(1)Gilbert综合征:最常见,国内发病率2%,酶活性为正常水平的25%~50%,症状轻,胆红素:17~102umol/L,对苯巴比妥有反应,感染、饥饿、NSAIDs等药物可诱发黄疸反复。整体病程表现为良性、慢性、反复发作的轻度黄疸。
(2)Crigler-Najjar综合征型Ⅱ型:全球发病率1/100万,为罕见病,酶活性约为正常水平的10%,中度黄疸,胆红素102~342umol/L。
(3)Crigler-Najjar综合征Ⅰ型:极为罕见,酶活性<正常水平1%,重度黄疸,胆红素342~769umol/L,一般在生后数日新生儿期即出现重症黄疸,常合并核黄疸,需血浆置换、尽早确诊、尽早肝移植。一般预后差,多于2岁内死亡。