
一、前沿聚焦,跨域协作共克儿童生长难题
2025年6月24日,上海儿童医学中心与深圳市人民医院联合举办儿童生长发育学术研讨会。本次会议聚焦五大疑难病例,通过腾讯会议平台集结沪深两地顶级专家,围绕体格生长和智能发育与内分泌遗传代谢的关系,从基因诊断瓶颈、罕见病管理、生育力保存等核心议题展开深度探讨,为儿童生长发育疾病的精准诊疗提供新范式。

1. 女童,性早熟、发育迟缓伴复杂基因变异: 36+6剖宫产、臀位、2120g、47cm,头围33cm,无窒息抢救史。外院产前诊断:染色体4p16.1p15.33区段检出638Kb微重复;孕32+4周B超:臀位,脐带绕颈一周,胎儿大小相当于29周1天,胎儿体重<P10。染色体:46, XX, lqh+, 16qh+。Trio-WES:PPP3CA:c.1069G>T,p.V357F(de novo)。青春期早发育、智力发育落后,先天性髋关节发育不良术后。家系父母表型正常。基因检测报告提示PPP3CA变异为疑似致病。
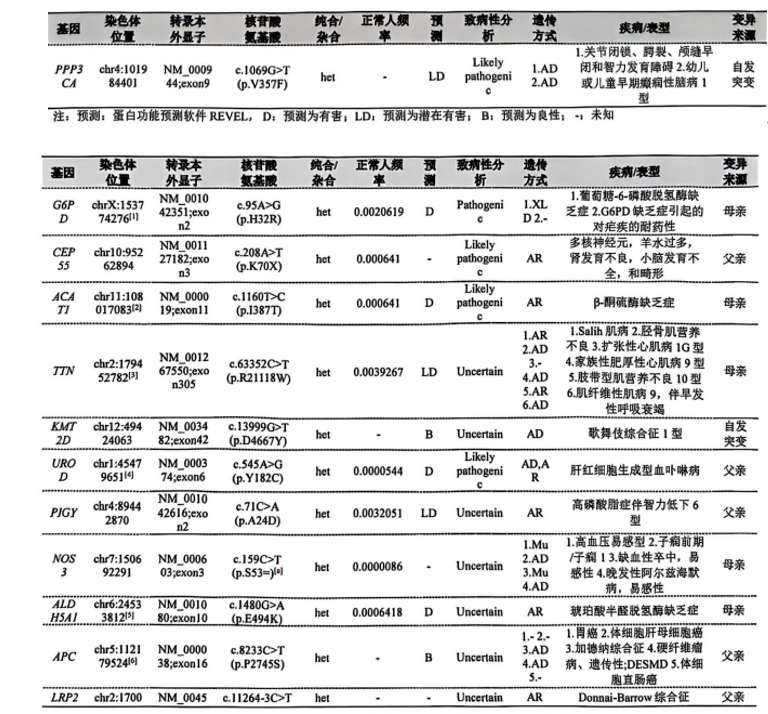
分享:1、基因检测报告“其他变异“列表KMT2D:c.13999G>T,p.D4667Y变异位于KMT2D基因43号外显子的最后一位碱基,该变异为新发变异,软件预测可能会影响KMT2D基因剪接,结合患者临床表现以及面容特征,提示可能为Kabuki综合征,进一步进行外周血RNA测序,验证KMT2D变异的致病性。2、补充头颅X光/CT(排查颅缝早闭)、眼科会诊(视网膜脱离风险)、脑电图(监测癫痫)。3、全面的诊断性神经发育评估。4、二胎生育前需排除生殖腺嵌合风险,孕期羊水检测谨慎解读嵌合比例。
小结:母亲孕期产前保健的任何的异常,都可能隐藏着我们尚未理解和发现的病理情况,会随着患儿的成长逐渐表现,我们需要对产前保健异常信息母子长期追踪,建立生物学信息与表型之间因果关系的数据库,整合产科、新生儿科、儿科直至成人的健康信息,提高对疾病因果的认识。同时需要根据患儿成长中逐渐出现的表型,重新分析基因数据和对数据库进行补充、更新。同时根据患儿即时的神经行为发育进行个性化定制的诊疗方案进行功能改善。
2.嵌合型特纳综合征伴性早熟:Turner综合征,青春期早发育(快进展),身材矮小。染色体核型:45,X(12)/46,XX(38)。

分享:1、生育力保存:卵巢组织冻存黄金窗口期为骨龄≤10岁且AMH>1.1 ng/ml;AMH<0.5 ng/ml时启动紧急冻存。2、性早熟治疗:亮丙瑞林+生长激素延长生长周期。3、自然妊娠可能性:外周血46,XX比例≥30%且AMH≥1.0 ng/ml时可尝试,但自发流产率>60%。4、数据支持:冻存卵巢组织移植后活产率28%(《Fertil Steril》2021)。
小结:绝大部分Turner综合征(TS)的患者因先天性卵巢发育不全而无法正常性发育和生育,但部分嵌合型患者存在性发育和生育可能。青春期发育过早对TS患儿的身高和生活质量影响,与生育力保护形成矛盾,把握适合的治疗节点是提高患儿生存质量的关键。
3. 营养不良伴发育落后(父源KMT2E变异):9月体检发现消瘦/营养不良(体重/身长<P3),预警征阳性,血氨、血乳酸增高、颅脑平扫:双侧额叶脑外间隙增宽,胎儿至儿童期生长发育监测(深圳市人民医院研发)提示胎儿期减速生长,双顶径、头围均<P10。WES:1、HIBCH,NM_014362.3:c.763C>T(p.Arg255*),杂合,母源;2、KMT2E, NM_182931.2:c.1348T>C(p.Tyr450His),杂合,父源。家系:父母表型正常。经干预贫血、营养不良纠正,血氨、血乳酸持续增高,发育行为症状无明显加重。
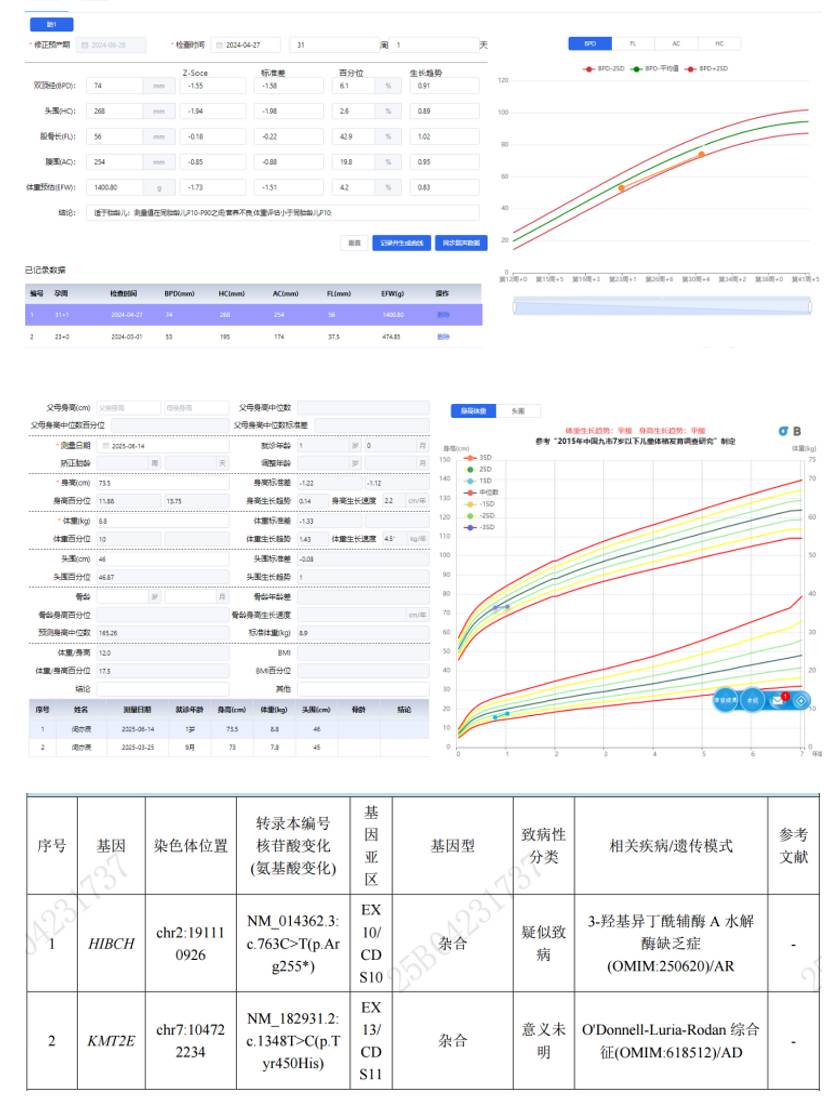
小结:胎儿期生长减速需要关注,往往提示胎儿存在病理性因素,患儿生后出现发育问题伴喂养困难、营养不良、乳酸、血氨升高,注意排除有机酸代谢问题,必要时除基因检测也需要串联质谱检查。继续关注患儿生长发育监测,对临床表现的变化及时掌握,必要时重分析基因数据,根据临床变化重新进行全面评估并制定个性化干预方案。
4. 矮小症、青春期早发育伴皮肤咖啡斑:身材矮小、身高增长缓慢、青春期早发育、皮肤咖啡斑、亚临床甲状腺功能亢进,高甘油三酯血症,WES未见异常。

分享:1、优先皮肤活检,重点分析是否存在NF1、GNAS等基因的体细胞嵌合变异。2、鉴别诊断:补充骨扫描、垂体MRI。3、若为体细胞嵌合:同胞再发风险<1%,若为NF1/PTEN种系突变:常显遗传(50%风险)。4、治疗:明确诊断后再考虑是否适合生长激素治疗,对高甘油三酯饮食干预。
小结:McCune-Albright综合征(MAS)虽然常见于女性患者但男性患者仍然可见,患儿仍需排查此综合征。其次,嵌合比例、受检样本、测序深度、测序覆盖度等因素可影响嵌合变异的检出,因此基因检测阴性仍不能排除NF1,尤其是节段性神经纤维瘤。需要注意节段性神经纤维瘤患者精子细胞也可能携带NF1变异,因此建议在生育后代时进行产前诊断。
5. 矮小伴全面发育迟缓,基因检测阴性:胎儿期发现股骨短小,婴幼儿期诊断全面发育迟缓进行干预,体格检查生长发育严重落后(体重19k、-2.0 sd;身高110.2 cm、<-3.0sd;BMI 15.7、-0.1 sd; 头围47.0cm、<-2.0sd)。WES:1、SLC7A7,NM_001126106.2:c.625+1G>A;2、NSMCE2,NM_173685.2:c.217C>T(p.Arg73Trp);3、TBCD,NM_005993.4:c.490C>T(p.Pro164Ser)。家系:父母表型正常。
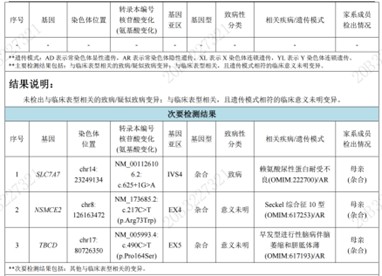
小结:未发现与表型明确相关的致病变异,提示几种可能:表型相关基因未覆盖、存在复杂遗传机制、或临床表型描述不充分。报告未提供具体临床表现是个重大缺陷。基因检查为我们探索疾病发生提供了一个有益的途径,但基因技术与临床诊疗的匹配仍需要漫长的融合,需要医患不断补充和完善信息并建立长期随访的因果联系。
三、活动总结
1、技术创新有效的应用于临床工作。例如上海交通大学医学院附属上海儿童医学中心王秀敏教授团队"罕见病面部识别系统",深圳市人民医院韩煊医生团队“胎儿至儿童期生长发育监测系统”,提高了临床诊疗水平。
2、遗传检测技术深入应用,例如RNA-Seq技术捕获深度内含子变异,动态重分析新表型数据。另外,完善罕见病诊疗的标准化临床路径,如特纳综合征患者生育力保存评估和节段性神经纤维瘤患者配子携带者NF1发生可能进行遗传咨询。
3、根据金星明教授提出"三维联动"模型(临床联合会诊+多中心基因数据库+生育力保存技术转化),规范发育行为学诊断,建立跨学科协作。
